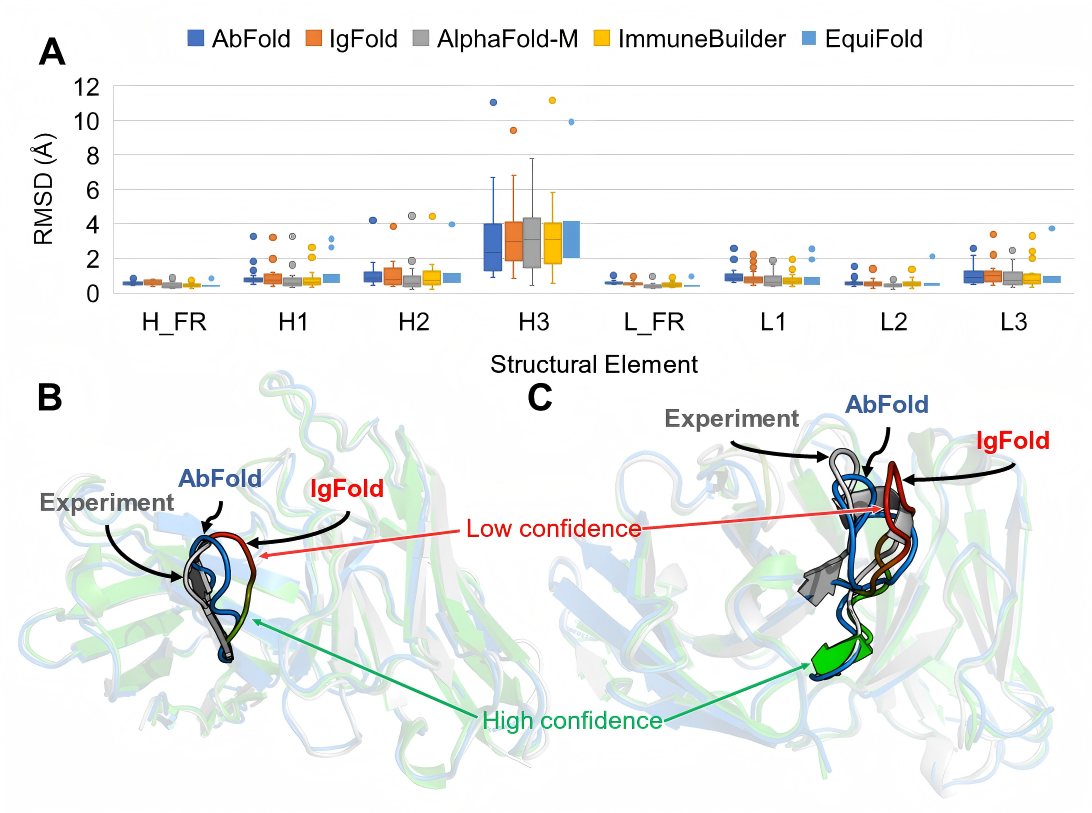
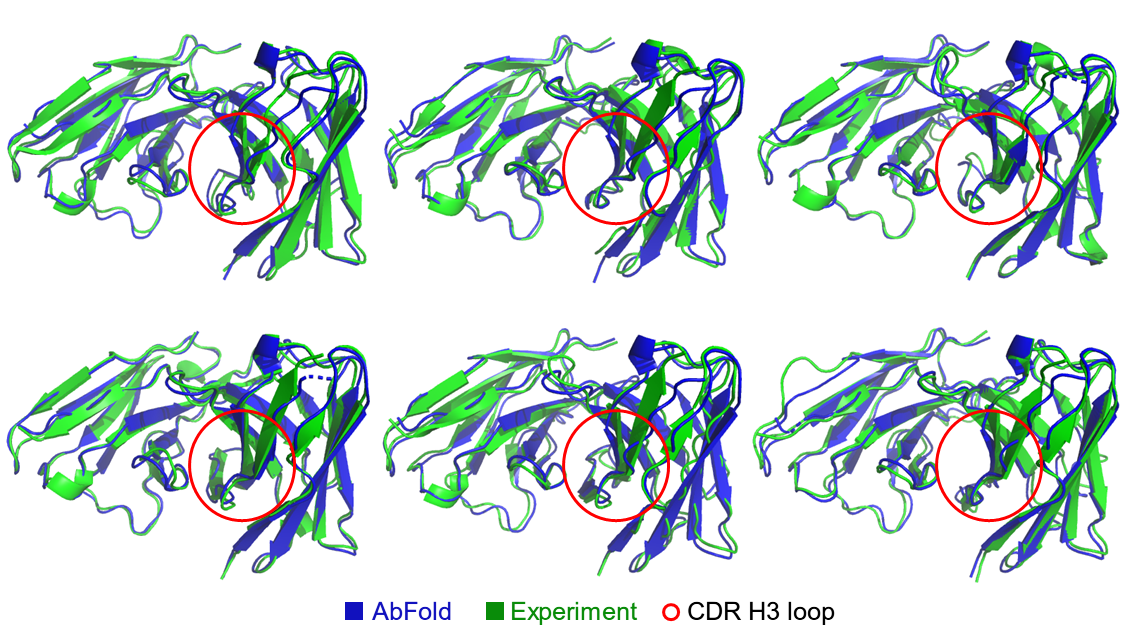
Achieve Full-Atomic Precision Prediction of Antibody Structures
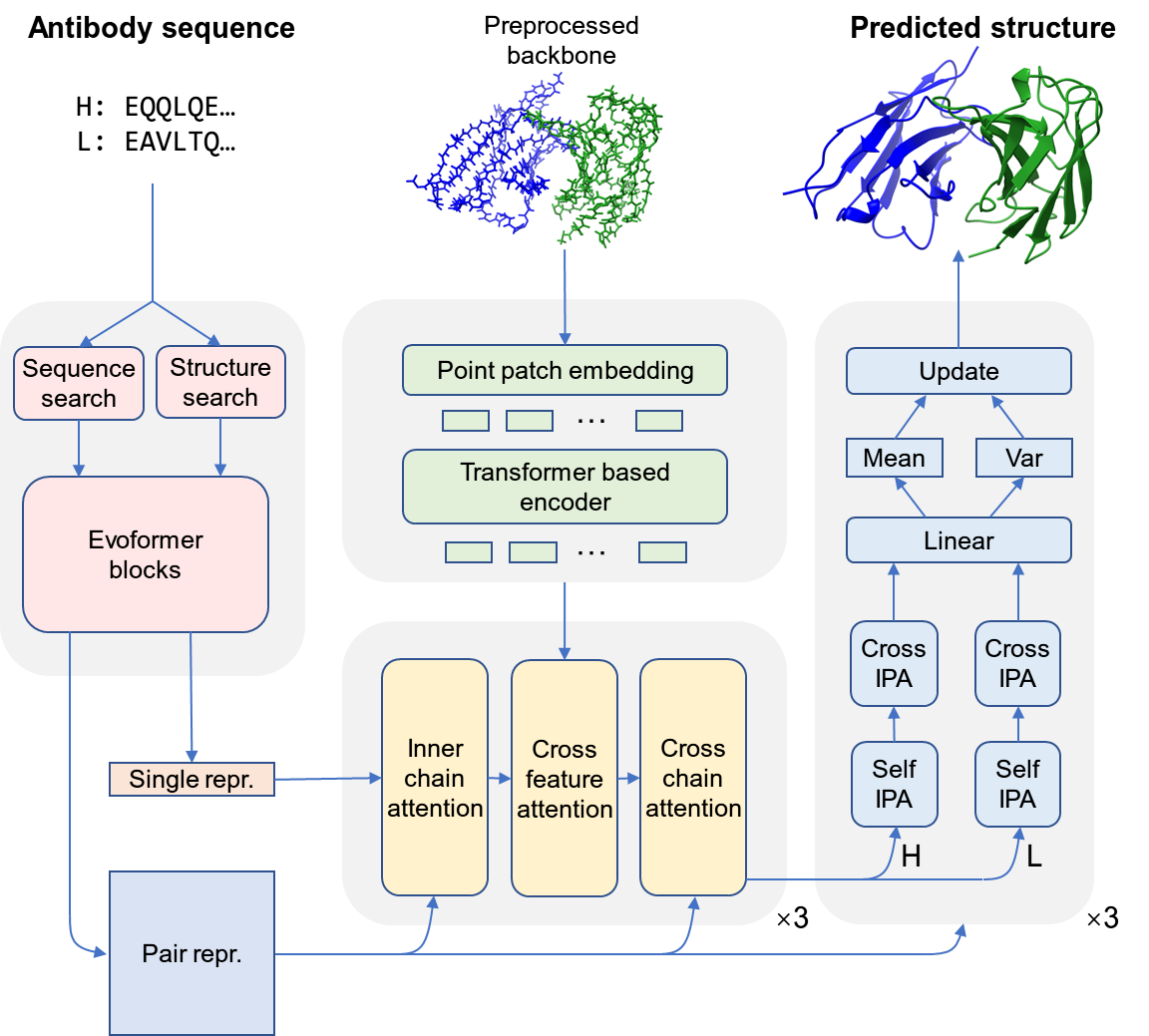
AbFold enables end-to-end modeling of antibody conformations with full-atomic precision. First, it uses MMseqs2 to search the Uniref90 sequence database to obtain homologous sequences of the antibody sequence and their multiple sequence alignments (MSAs), and extracts language model features via the antibody language model AntiBERTy. Then, transfer learning is employed to extract homologous sequence features and language model features from AlphaFold-Multimer and IgFold, respectively. Subsequently, these two types of features are fused through an information fusion module based on fully connected layers to generate the final sequence features, which are then input into a neural network based on a variational autoencoder for dynamic modeling of antibody structures.